INTRODUCTION
Acid mine drainage (AMD) is a persistent legacy of mining activities, characterized by highly acidic water enriched with dissolved metals and sulfate [1,2]. When sulfide minerals are exposed to oxygen and water, oxidation reactions generate acidity and mobilize metals that can contaminate streams and groundwater for years or even decades [3,1]. The resulting water often exhibits very low pH, elevated Fe and other metals, high total dissolved solids, and visible turbidity, creating acute and chronic risks to aquatic ecosystems and downstream users [1,2].
The urgency to treat AMD is driven not only by environmental compliance but also by the need to protect water resources and reduce long-term remediation costs [4,5]. Untreated AMD can degrade habitats, disrupt biological communities, and corrode infrastructure, while persistent metal loading can render water bodies unfit for agriculture or domestic use [4,5]. These impacts are particularly significant in regions where mining is a major economic activity and local communities rely on surface water for daily needs [1].
Conventional AMD treatment relies on several well established approaches, each with distinct limitations. Chemical precipitation using lime or limestone is widely applied for neutralization and metal removal [6]. It nevertheless generates large volumes of sludge, requires continuous chemical dosing, and is ineffective for sulfate reduction below gypsum solubility. Discharge limits for sulfate are often difficult to meet as a result [7,8]. Biological methods include passive systems (e.g., constructed wetlands) and active sulfate-reducing bioreactors. They can remove metals and sulfate with minimal chemicals but are typically slow, sensitive to influent quality, and may require external carbon sources [3]. Adsorption and ion exchange offer good removal of specific contaminants but entail recurring media or regenerant costs and disposal of spent materials [1,2]. Membrane processes such as reverse osmosis can produce high-quality effluent but are energy-intensive and prone to fouling; the concentrated brines they produce need further management [1]. These drawbacks have motivated the search for compact, chemical-free or low-chemical alternatives that can achieve robust removal and stable effluent quality [9,6].
Electrocoagulation (EC) has gained attention as a compact and efficient process for AMD treatment because it generates coagulant species in situ, promotes rapid metal precipitation, and elevates pH without external chemical dosing [10,11,12,6]. Laboratory and field studies have demonstrated high removal of heavy metals (e.g., Fe, Zn, Mn, Cu) from real and synthetic AMD, with reported efficiencies often exceeding 90% under optimized conditions, supporting its potential for scaling and further development [10,12,13,11]. However, EC alone can leave residual dissolved ions, fine colloids, or elevated TDS, especially when influent chemistry is complex. Integrating EC with adsorption or filtration media (e.g., silica sand, activated carbon, zeolite) offers a logical hybrid strategy. EC performs bulk removal and pH correction, while the downstream stage provides polishing to achieve more stable and compliant effluent quality [14,15,2].
Despite the promise of hybrid treatment trains, few studies have systematically evaluated EC coupled with low-cost adsorption/filtration media for AMD under practical operating windows. Reported work often focuses on single-unit performance or targets a limited set of parameters, leaving uncertainty about the combined effect on metal removal, TDS reduction, and final pH stabilization [7,8,16]. There remains a clear need to quantify the operational synergy of EC and adsorption stages and to identify conditions that maximize overall treatment efficiency.
Therefore, this study investigates a hybrid EC–adsorption/filtration system for AMD treatment using aluminum electrodes and low-cost media (silica sand, activated carbon, and zeolite). The effects of applied voltage and treatment time are evaluated, and performance is quantified in terms of Fe removal, TDS reduction, and pH adjustment. The work provides a practical assessment of operational conditions and highlights the role of a polishing stage in achieving compliant effluent quality.
MATERIALS AND METHODS
Materials
Acid mine drainage (AMD) samples were collected from a mine drainage channel and stored in high-density polyethylene containers at 4 °C prior to use. Initial pH, Fe concentration, and TDS were measured to establish baseline characteristics and treatment targets. Aluminum plate electrodes were used as both anode and cathode in the EC reactor. The adsorption/filtration media consisted of washed silica sand, commercial granular activated carbon, and natural zeolite, all pre-rinsed with deionized water and dried before use. pH was adjusted using NaOH and HCl solutions. Chemicals used for Fe analysis by the phenanthroline method included hydroxylamine hydrochloride, 1,10-phenanthroline, and ammonium acetate buffer. All reagents were analytical grade, and deionized water was used for dilutions and rinsing.
Experimental Setup
Electrocoagulation experiments were conducted in a 10 L batch reactor equipped with a DC power supply. The reactor was filled with 6 L of AMD sample, and aluminum electrodes were installed as anode and cathode and connected to the power supply using insulated clips.
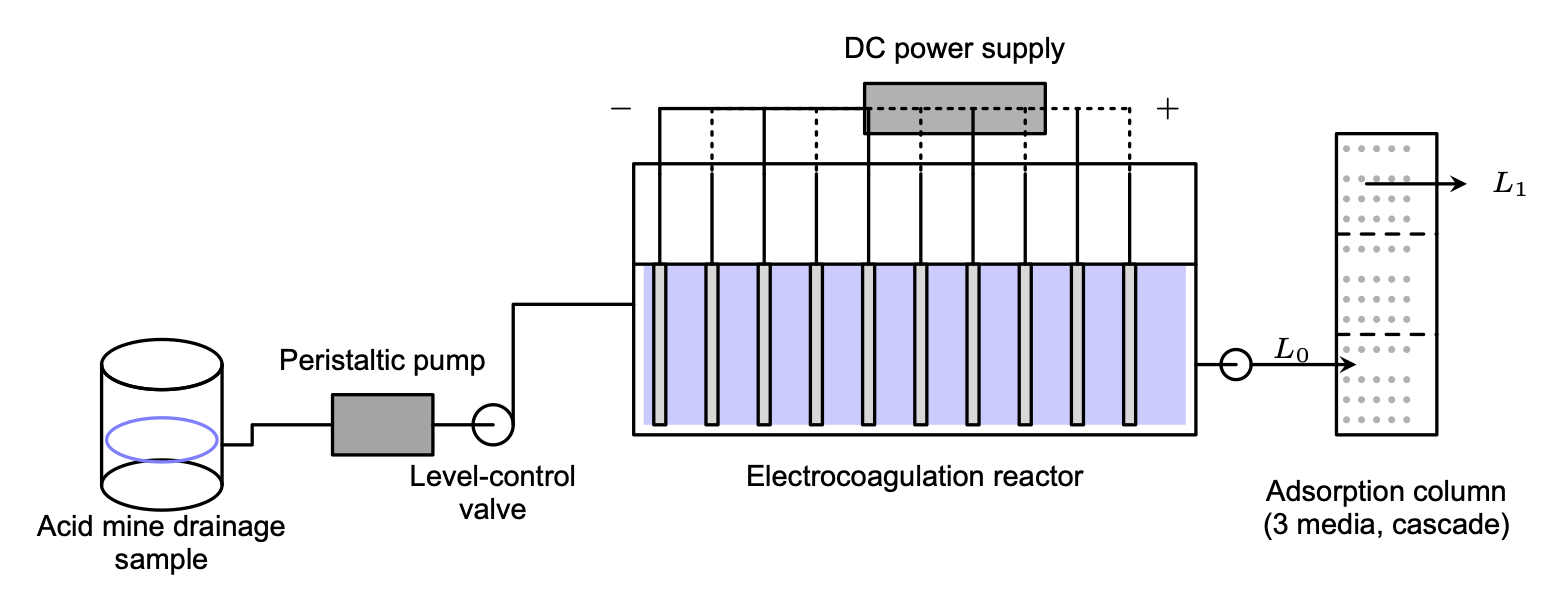
A schematic of the experimental setup is shown in Figure 1. AMD sample was pumped from a closed reservoir through a peristaltic pump and a level-control valve into the electrocoagulation reactor. The reactor contained ten parallel aluminum plate electrodes connected to a DC power supply in an alternating polarity arrangement. After the predetermined electrocoagulation contact time, the treated water exited the reactor and was passed through an adsorption column packed with three media (silica sand, activated carbon, and zeolite) in cascade before collection.
Figure 1. Schematic of the hybrid electrocoagulation–adsorption experimental setup. AMD sample is pumped from the reservoir through a peristaltic pump and level-control valve into the electrocoagulation reactor; DC power is supplied to alternating electrodes. Effluent is then passed through a three-media adsorption column (L0: inlet, L1: outlet).
Electrocoagulation Procedure
The electrocoagulation experiments were conducted in batch mode under different voltages and contact times. The applied voltage was varied at 1, 2, and 3 V with contact times of 10, 20, 30, 40, 50, 60, 90, and 120 min. At each predetermined time, a sufficient sample volume was withdrawn for analysis. pH, TDS, and Fe concentration were measured to evaluate EC performance.
Adsorption Procedure
After electrocoagulation, the treated AMD was directed to a multimedia filtration reactor packed in sequence with silica sand, activated carbon, and zeolite. Each medium was washed separately with clean water until the rinse water was clear to remove dust, fine particles, and organic impurities, then oven-dried at 105 °C. The dried media were sieved to obtain a uniform particle size distribution consistent with the filter design. The EC effluent was pumped through the filtration column at a measured flow rate, and the treated water exited via the outlet. Effluent samples were collected for Fe, pH, and TDS analyses.
Analytical Methods
pH and TDS were measured using a Milwaukee MW105 multiparameter meter. Prior to analysis, the instrument was calibrated with pH 4.00 and 7.00 buffer solutions for the pH probe and with a TDS standard solution for the conductivity/TDS probe. Samples were brought to room temperature and stirred gently before measurement; the probe was rinsed with deionized water between samples.
Fe concentration was determined by the 1,10-phenanthroline spectrophotometric method. An aliquot of the sample was transferred to a volumetric flask and diluted if necessary to bring the Fe concentration within the calibration range. Hydroxylamine hydrochloride solution was added to reduce Fe3+ to Fe2+, followed by 1,10-phenanthroline solution and ammonium acetate buffer to maintain pH 3–5 for complex formation. The mixture was allowed to stand for 10–15 min for full color development, and absorbance was measured at 510 nm using a spectrophotometer. A calibration curve was prepared from Fe standard solutions treated identically.
Data Analysis
Removal efficiency for Fe and TDS was calculated as:
where C0 is the initial concentration and C is the concentration after treatment. For the electrocoagulation stage, removal was evaluated as a function of applied voltage (1, 2, and 3 V) and contact time (10–120 min). For the hybrid system, removal was assessed before and after the adsorption step to quantify the polishing effect. pH was measured directly, and the change in pH (ΔpH) across the adsorption column was determined. Experiments were conducted in replicate; results are reported as mean ± standard deviation.
RESULTS AND DISCUSSION
Electrocoagulation Performance
Since the electrocoagulation process is a pH adjustment process, the pH evolution is the first and most important parameter to be monitored. Figure 2 shows the pH evolution during electrocoagulation at different voltages. The electrocoagulation process progressively increased AMD pH with contact time, and the rate of increase was strongly voltage-dependent. At 3 V, pH rose from 1.98 to 9.25 within 120 min, indicating rapid neutralization and substantial alkalinity generation. At 2 V, the pH increase was slower but still significant, reaching 6.00 after 120 min. The 1 V condition showed the weakest response, ending at pH 5.15 after 120 min. These trends demonstrate that higher voltages (and thus higher current density) accelerate electrochemical dissolution of the anode and cathodic water reduction, producing more hydroxide ions and increasing pH [17,18].
Figure 2. pH evolution during electrocoagulation at different voltages with standard deviation error bars.
The further more detailed analysis of the pH evolution shows the temporal profile that suggests two-phase evolution. An initial period (0–40 min) with modest pH change, followed by a sharper rise between 50–120 min, especially at 3 V. This pattern is consistent with the accumulation of alkalinity and the onset of the metal hydrolysis/precipitation reactions that consume acidity. The increasing pH likely promotes Al(OH)3 formation and sweep flocculation, which is central to the AMD treatment process [19].
The increase in pH can be supported by the following electrochemical and hydrolysis reactions:
From an operational perspective, 3 V achieves a near-neutral to alkaline pH (pH ≥ 7) after 90 min, while 2 V requires longer contact time to approach neutrality, and 1 V may be insufficient for complete neutralization within the tested time. This indicates a trade-off between energy input (voltage) and treatment time [20,6]. The higher the voltage, the shorter the treatment time, but the higher the energy consumption.
Figure 3 presents Fe removal efficiency over time at different voltages. Fe removal increased monotonically with contact time for all voltages, with a strong dependence on applied voltage. At 3 V, removal rose rapidly from 0% at 0 min to 23.6% (40 min), 37.2% (60 min), and reached 82.5% at 120 min. The 2 V condition achieved moderate performance, reaching 26.0% at 60 min and 61.9% at 120 min. The 1 V run showed the slowest kinetics, with only 15.3% at 60 min and 39.1% at 120 min. These trends indicate that higher voltage (and thus higher current density) accelerates Fe removal, consistent with increased Al dissolution and faster formation of Al(OH)3 flocs that promote sweep coagulation and coprecipitation [6,11]. The low standard deviations (typically 1–4%) suggest good repeatability of the removal profiles across replicates.
Figure 3. Fe removal during electrocoagulation at different voltages with standard deviation error bars.
The removal-time profiles suggest a two-stage behavior: an initial slow period (0–40 min) followed by a sharper increase after 50 min, especially at 3 V. This inflection coincides with the pH rise observed in Figure 2, indicating that once the system approaches near-neutral pH, Fe(III) hydrolysis and precipitation become dominant, enhancing removal [9,12]. Practically, 3 V achieves high Fe removal within 90–120 min, while 2 V and 1 V require longer contact times and still show lower final efficiencies, highlighting the trade-off between energy input and treatment time for achieving target Fe removal.
Compared with the literature, Fe removal efficiencies in AMD and mine waters are often higher under optimized or higher-current conditions. For example, Alam et al. reported up to 99.95% Fe removal from iron-ore mine wastewater by electrocoagulation under optimized electrolyte and voltage conditions [10]. Foudhaili et al. achieved >94% Fe removal at 60 min using Fe electrodes in Fe-rich synthetic AMD [13], and Stylianou et al. reported 99.9% Fe removal from real AMD using Al electrodes at 20 mA/cm2 [12]. In comparison, our maximum Fe removal of 82.5% at 120 min (3 V) is lower, which is likely attributable to the lower applied voltage/current density, differences in initial Fe loading and matrix composition, and the more gradual pH rise in our system. These comparisons indicate that higher current densities and faster pH neutralization generally yield higher Fe removal, but at the expense of greater energy input.
Figure 4 shows TDS removal efficiency over time at different voltages. TDS removal increased with contact time and voltage, indicating improved ionic and colloidal removal at higher current density. At 3 V, removal rose from 0% at 0 min to 32.9% at 40 min and reached 68.9% at 120 min. At 2 V, the increase was more gradual, reaching 21.8% at 60 min and 50.6% at 120 min. The 1 V condition showed limited performance with only 25.0% removal at 120 min, and a small fluctuation around 50–60 min, which is likely within experimental variability. The low standard deviations (typically <4%) support the reproducibility of these trends.
The stronger TDS reduction at higher voltage is consistent with enhanced coagulant generation and more efficient removal of dissolved and fine suspended species via sweep coagulation, adsorption onto Al(OH)3 flocs, and subsequent settling [11,21]. From an operational perspective, 3 V provides substantial TDS reduction within 90–120 min, whereas 2 V and especially 1 V would require longer contact times or additional polishing to reach comparable levels.
Figure 4. TDS removal during electrocoagulation at different voltages with standard deviation error bars.
Adsorption Polishing Performance
The adsorption stage serves as a polishing step to remove residual metals and sulfate that remain after electrocoagulation. Since different electrocoagulation voltages produce different effluent pH values, the adsorption step was performed under three distinct initial pH conditions corresponding to the post-EC effluent: pH 9.25 (from 3 V EC), pH 7.00 (from 2 V EC), and pH 5.30 (from 1 V EC). Figure 5 shows the pH comparison before and after adsorption for these three initial conditions.
At an initial pH of 9.25 (post-3 V EC effluent), the pH decreased to 8.70 ± 0.28 after adsorption (ΔpH = −0.55). Similarly, at an initial pH of 7.00 (post-2 V EC effluent), the pH dropped slightly to 6.80 ± 0.21 (ΔpH = −0.20). In contrast, at an initial pH of 5.30 (post-1 V EC effluent), the pH increased to 5.90 ± 0.21 (ΔpH = +0.60).
These trends indicate that the direction of pH change during adsorption is governed by the initial pH condition. Under alkaline to neutral conditions (pH 7–9), adsorption involves proton release via ion-exchange or surface complexation mechanisms, resulting in a net pH decrease. Under acidic conditions (pH < 6), the adsorbent surface may protonate or buffer residual acidity, leading to a net pH increase. This pH-dependent behavior is consistent with the amphoteric nature of typical adsorbent surfaces [15,22] and highlights the importance of electrocoagulation pretreatment in conditioning the effluent pH for optimal adsorption performance.
From a practical perspective, the final pH values after adsorption remain within acceptable discharge limits (pH 6–9) [1,4] for the initially alkaline (8.70) and neutral (6.80) conditions, while the initially acidic condition (5.90) approaches the lower regulatory threshold. These results demonstrate that higher electrocoagulation voltage not only improves metal removal during EC but also produces favorable initial pH conditions that ensure the combined EC–adsorption system yields effluent suitable for direct discharge.
Figure 5. pH values before (post-EC) and after adsorption polishing at three initial pH conditions corresponding to different EC voltages. Error bars represent standard deviation.
Figure 6 shows Fe removal before and after adsorption polishing at the three initial pH conditions. At an initial pH of 9.25 (post-3 V EC), Fe removal increased from 82.53 ± 1.52% to 90.03 ± 0.76%, an additional removal of 7.50 percentage points. At an initial pH of 7.00 (post-2 V EC), removal rose from 61.90 ± 4.17% to 71.81 ± 2.27% (+9.91 percentage points). At an initial pH of 5.30 (post-1 V EC), removal increased from 39.12 ± 1.52% to 53.32 ± 2.65% (+14.20 percentage points).
Although the absolute increase in Fe removal was highest under acidic conditions (+14.20%), the efficiency of removing residual Fe was greatest at high pH. At initial pH 9.25, adsorption removed 43% of the remaining Fe (7.50/17.47), compared to 26% at pH 7.00 (9.91/38.10) and 23% at pH 5.30 (14.20/60.88). This enhanced efficiency at alkaline pH is consistent with the formation of Fe(OH)3 precipitates that are more readily adsorbed, as well as reduced competition from H+ ions for active adsorption sites [9,23].
From a practical standpoint, only the high-pH condition (post-3 V EC) achieved Fe removal exceeding 90%, approaching typical discharge targets [1]. The neutral condition (post-2 V EC) reached 71.81%, while the acidic condition (post-1 V EC) attained only 53.32%. These results underscore the synergistic benefit of combining high-voltage electrocoagulation with adsorption polishing: the EC stage raises pH and removes bulk Fe, creating favorable conditions for efficient residual Fe capture during adsorption [7].
Figure 6. Fe removal before (post-EC) and after adsorption polishing at three initial pH conditions corresponding to different EC voltages. Error bars represent standard deviation.
Figure 7 presents TDS removal before and after adsorption polishing at the three initial pH conditions. At an initial pH of 9.25 (post-3 V EC), TDS removal increased from 68.93 ± 1.60% to 76.53 ± 0.46%, an additional removal of 7.60 percentage points. At an initial pH of 7.00 (post-2 V EC), removal rose from 50.58 ± 3.56% to 60.63 ± 1.28% (+10.05 percentage points). At an initial pH of 5.30 (post-1 V EC), removal increased from 24.96 ± 0.04% to 35.29 ± 0.04% (+10.33 percentage points).
Similar to Fe removal, the efficiency of removing residual TDS was highest at alkaline pH. At initial pH 9.25, adsorption removed 24.5% of the remaining TDS (7.60/31.07), compared to 20.3% at pH 7.00 (10.05/49.42) and 13.8% at pH 5.30 (10.33/75.04). This pattern is consistent with enhanced coprecipitation and favorable adsorbent surface charge at higher pH, which promotes capture of dissolved ionic species [24,20]. Notably, the very low standard deviations at the 1 V condition (<0.05%) indicate excellent experimental repeatability, even though overall removal remained limited.
From a practical perspective, the combined EC–adsorption system achieved 76.53% TDS removal under optimal conditions (post-3 V EC), representing a substantial reduction in dissolved solids. The neutral and acidic conditions yielded 60.63% and 35.29% removal, respectively, indicating that higher electrocoagulation voltage is essential to maximize both bulk and residual TDS removal.
Figure 7. TDS removal before (post-EC) and after adsorption polishing at three initial pH conditions corresponding to different EC voltages. Error bars represent standard deviation.
Operational Optimization
The experimental results demonstrate a clear trade-off between applied voltage and treatment time in the electrocoagulation stage [25,20]. Higher voltage accelerates pH neutralization and contaminant removal but presumably increases energy consumption, while lower voltage conserves energy at the cost of extended treatment time and reduced removal efficiency.
Increasing the applied voltage from 1 V to 3 V significantly improved all treatment outcomes. At 120 min, pH rose from 5.15 (1 V) to 9.25 (3 V), Fe removal increased from 39.1% to 82.5%, and TDS removal improved from 25.0% to 68.9%. This enhancement is attributed to higher current density at elevated voltage, which accelerates anodic aluminum dissolution and cathodic hydroxide generation. The threefold increase in voltage yielded approximately twofold improvements in Fe and TDS removal, suggesting diminishing returns at higher voltages that should be balanced against energy costs.
The time-series data reveal two distinct phases in pH evolution and contaminant removal. During the initial 40 min, changes were modest across all voltages, likely reflecting the accumulation of coagulant species and gradual pH adjustment. After 50 min, removal rates accelerated markedly, particularly at 3 V, coinciding with the onset of significant metal hydroxide precipitation. This inflection point suggests that a minimum contact time of 60–90 min is required for effective treatment, with 120 min providing near-maximum removal under the tested conditions.
Based on the combined electrocoagulation–adsorption results, the optimal operating condition identified in this study is 3 V for 120 min electrocoagulation followed by adsorption polishing. This configuration achieved final removal efficiencies of 90.0% for Fe and 76.5% for TDS, with effluent pH of 8.7—all within typical discharge standards. For applications where energy minimization is prioritized, 2 V operation may offer a reasonable compromise, achieving 71.8% Fe removal and 60.6% TDS removal after the combined treatment, though with reduced margins relative to discharge limits.
Mechanistic Insights
Based on the observed trends and established electrocoagulation theory, the following mechanisms are proposed to explain the treatment performance in this study.
The electrocoagulation process involves three primary mechanisms operating in sequence. First, anodic dissolution releases Al3+ ions into solution according to Equation (3), while cathodic water reduction generates hydroxide ions and hydrogen gas (Equation 2). As hydroxide accumulates, the solution pH rises progressively, consistent with the pH evolution observed in Figure 2 [19].
Second, the released Al3+ ions undergo hydrolysis to form aluminum hydroxide species, predominantly Al(OH)3 at near-neutral to alkaline pH (Equation 4). These amorphous hydroxide precipitates possess high surface area and positive surface charge under acidic to neutral conditions, enabling them to destabilize and aggregate suspended particles and dissolved contaminants through charge neutralization and sweep flocculation [10].
Third, Fe removal likely occurs through multiple pathways: (i) coprecipitation with Al(OH)3 flocs, (ii) oxidation of Fe2+ to Fe3+ followed by precipitation as Fe(OH)3 at elevated pH, and (iii) adsorption onto freshly formed hydroxide surfaces. The strong correlation between pH rise and Fe removal (Figures 2 and 3) supports hydroxide precipitation as the dominant mechanism, consistent with previous electrocoagulation studies on iron-rich AMD [13].
The adsorption stage removes residual contaminants through surface-mediated processes. The pH-dependent behavior observed in this study—where adsorption efficiency for residual Fe and TDS was highest at alkaline initial pH—is consistent with surface complexation and electrostatic attraction mechanisms. At higher pH, the adsorbent surface likely carries a net negative charge, favoring uptake of residual cationic metal species through inner-sphere or outer-sphere complexation. Additionally, ion exchange between surface functional groups (e.g., –OH, –COOH) and dissolved metal ions may contribute to removal [14].
The slight pH decrease observed during adsorption at alkaline conditions (ΔpH = −0.20 to −0.55) is consistent with proton release accompanying ligand exchange or surface complexation reactions [22,14]. Conversely, the pH increase at acidic conditions (ΔpH = +0.60) suggests proton consumption or hydroxide release from the adsorbent surface, indicating buffering capacity under low-pH conditions [15].
The combined electrocoagulation–adsorption system exhibits synergy through sequential conditioning [7,21]. Electrocoagulation raises pH, removes bulk contaminants, and generates hydroxide flocs that may remain in suspension or settle. The subsequent adsorption step captures residual dissolved species under favorable pH conditions established by the EC pretreatment. This sequential approach achieves higher overall removal than either process alone would likely achieve, as the EC stage reduces contaminant loading and optimizes solution chemistry for efficient adsorption [26,8].
CONCLUSION
A hybrid electrocoagulation–adsorption system was evaluated for treating acid mine drainage. Electrocoagulation at 3 V for 120 min raised AMD pH from 1.98 to 9.25 and achieved 82.5% Fe removal and 68.9% TDS removal. Subsequent adsorption polishing increased total removal to 90.0% for Fe and 76.5% for TDS, with final effluent pH of 8.7—meeting typical discharge standards. Higher electrocoagulation voltage produced more favorable initial pH conditions for adsorption, enhancing residual contaminant capture efficiency. The combined system demonstrates synergistic benefits: electrocoagulation removes bulk contaminants and conditions solution pH, while adsorption polishes residual species. These results indicate that hybrid electrocoagulation–adsorption is a promising approach for AMD treatment, particularly where high removal efficiency and near-neutral effluent pH are required.